z-fasta: Indexing FASTA 17x Faster, and All the Things It Still Cannot Do
TLDR: z-fasta indexes FASTA files 9-17x faster than samtools while producing byte-identical .fai output. It is a zero-dependency static binary written in Zig. It handles 20/20 edge cases correctly. It has a streaming mode that uses 4 MB of RAM.
Why this exists
samtools faidx is the standard. It works. It is correct. It is also slow.
On a 3 GB human genome, samtools faidx takes 9.2 seconds on warm cache. Run it once and you do not notice. Run it in a pipeline that indexes hundreds of files and you wait.
I had been learning Zig for a few months. The language's strengths (no hidden control flow, explicit memory, direct SIMD) map directly onto the problem. A FASTA indexer is not complex. Scan bytes for lines starting with >, record offsets, write them out. The bottleneck is how fast you move bytes from disk to CPU.
I wanted to see how close to the hardware limit I could get.
What z-fasta does
z-fasta is a drop-in replacement for samtools faidx. It emits byte-identical .fai output. It also writes .zfi, a compact binary index for programmatic use.
zig build -Doptimize=ReleaseFast
# Emit samtools-compatible .fai to stdout
z-fasta index --emit-fai genome.fa > genome.fai
# Or create a binary .zfi index
z-fasta index genome.faThree modes:
- Default: mmap + SIMD scanning, with duplicate header detection.
- No-dedup: mmap + SIMD, no duplicate tracking. Fastest. Use when you trust your input.
- Low-memory: chunked reader, 4 MB buffer. For constrained machines where mmap is not available.
Performance
Benchmarked against samtools, seqkit (Go), and fastahack (C++) on three real datasets. All tests on an AMD Ryzen 9 3950X with warm cache, using hyperfine.
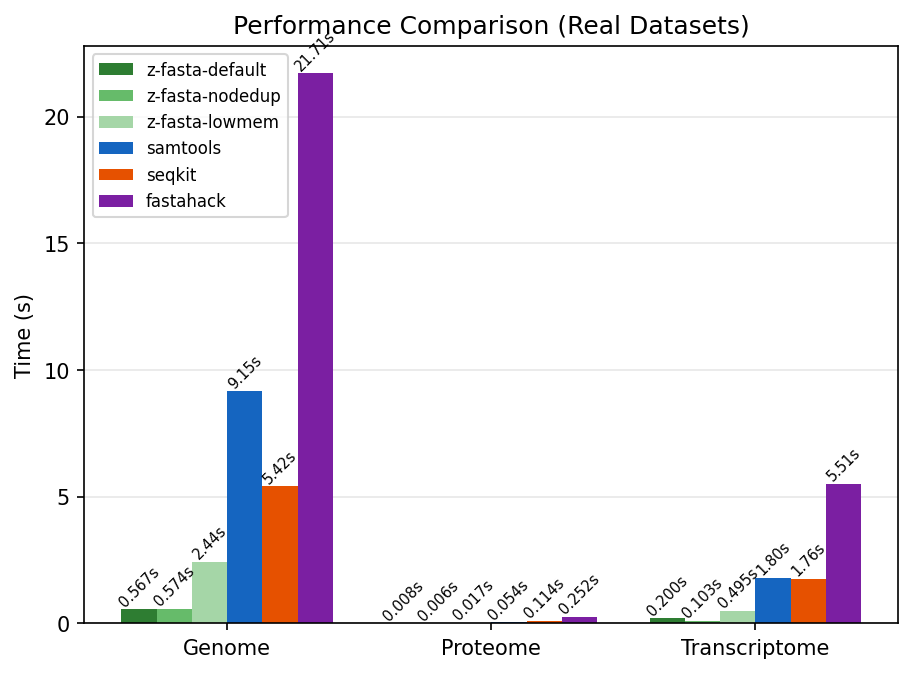
Indexing time in seconds across genome (3.0 GB), proteome (66 MB), and transcriptome (972 MB) datasets. Lower is better.
| Dataset | Size | z-fasta (no-dedup) | samtools | seqkit | fastahack | Speedup vs samtools |
|---|---|---|---|---|---|---|
| :--- | ---: | ---: | ---: | ---: | ---: | ---: |
| Genome | 3.0 GB | 0.57s | 9.15s | 5.42s | 21.71s | 16.1x |
| Transcriptome | 972 MB | 0.10s | 1.79s | 1.76s | 5.51s | 17.5x |
| Proteome | 66 MB | 0.006s | 0.05s | 0.11s | 0.25s | 9.4x |
z-fasta is faster than every tool on every dataset. The gap widens with file size.
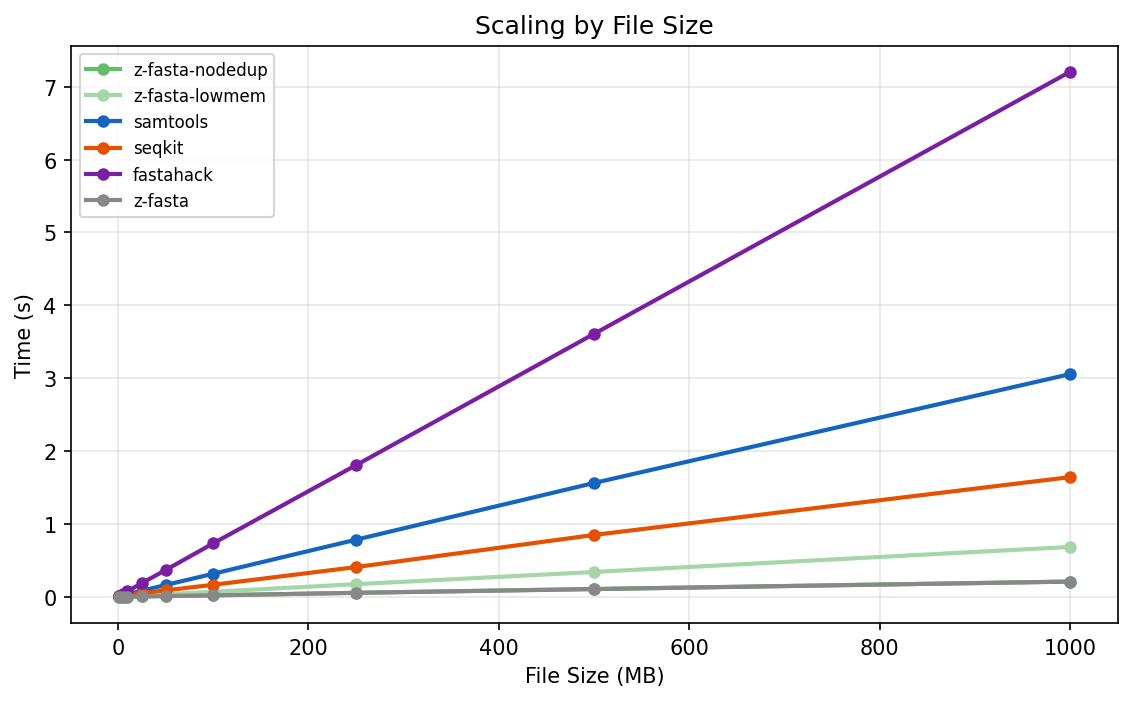
Indexing time vs file size. z-fasta and samtools both scale linearly. z-fasta’s slope is an order of magnitude shallower.
z-fasta and samtools both scale linearly with file size. z-fasta's slope is an order of magnitude shallower. At 1 GB, samtools takes 3 seconds. z-fasta takes 0.2 seconds.
Sequence count has almost no effect. At 100,000 sequences, z-fasta takes 0.02 seconds in no-dedup mode. The work is I/O-bound, not header-bound.
How it works
SIMD newline scanning
A FASTA indexer spends almost all of its time looking for \n. z-fasta uses Zig's @Vector types to scan 32 bytes at a time. On x86_64 this compiles to AVX2 vector compares. A 3 GB genome is one pass at memory bandwidth.
mmap by default
z-fasta memory-maps the entire file. The OS handles buffering. The CPU sees a flat byte array. No read() calls in userspace. No buffer management.
The tradeoff is that time reports VmRSS equal to the file size. The OS maps the file to virtual memory and time counts it
◆
The working set during indexing is a fraction of what VmRSS reports. The OS does not actually read the whole file into RAM.
. Actual private heap allocation is small: roughly 45 MB for the header hash map in default mode, under 1 MB in no-dedup mode.
If you cannot afford even the virtual memory footprint, --low-mem switches to a chunked reader with a 4 MB buffer. It is 3-4x slower than mmap but uses essentially no memory.
| Mode | Time (Genome) | Heap | RSS (reported) |
|---|---|---|---|
| :--- | ---: | ---: | ---: |
| no-dedup | 0.57s | < 1 MB | ~3 GB (mmap) |
| default | 0.57s | ~45 MB | ~3 GB (mmap) |
| low-mem | 2.44s | 4 MB | 4 MB |
| samtools | 9.15s | ~3 MB | ~3 MB |
Correctness
Speed means nothing if the output is wrong. I tested z-fasta against samtools on 20 edge cases: zero-byte files, missing trailing newlines, mixed \r\n endings, unicode headers, binary garbage mid-file, tab characters in sequence names, sequences with no line wrapping.
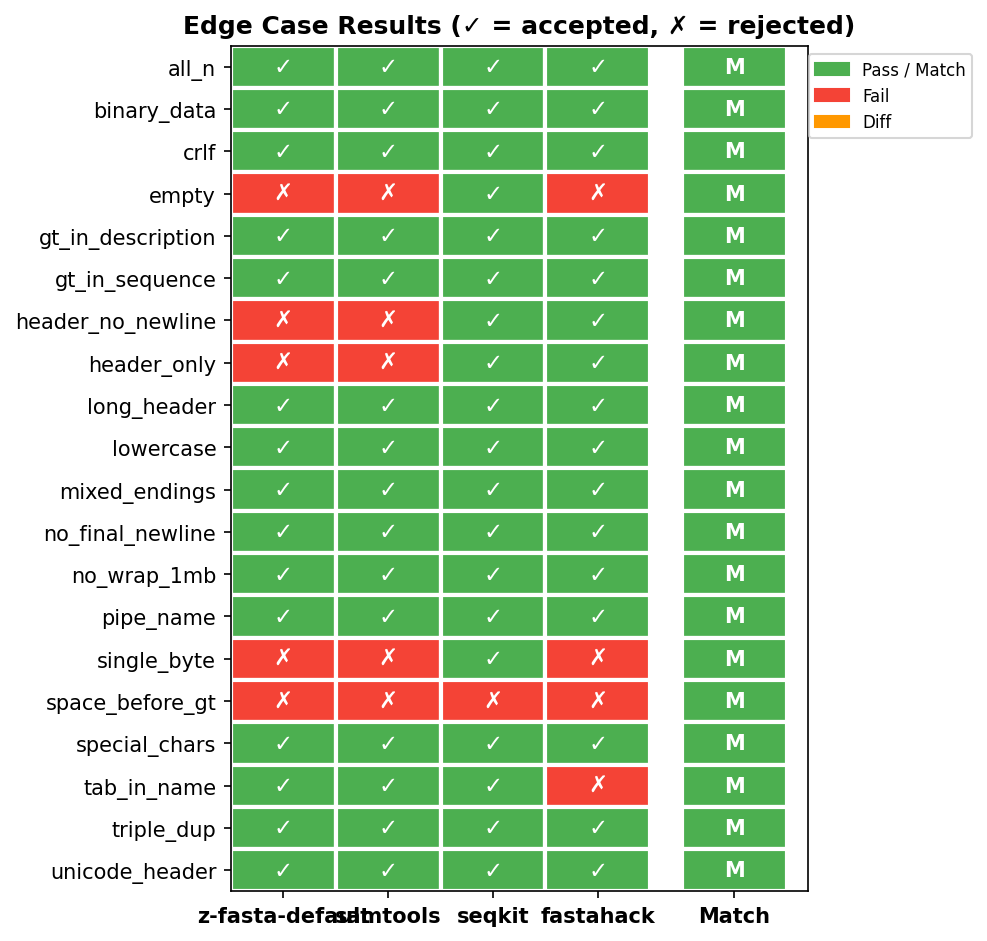
Edge case heatmap. Green = pass, red = fail. z-fasta matches samtools on all 20 cases, including exit codes for errors.
Result: 20/20 edge cases match samtools behavior exactly, including exit codes for error cases. seqkit silently accepts some malformed inputs that both samtools and z-fasta reject.
Honest Limitations
This is a proof of concept. It indexes FASTA and nothing else.
It was built with Zig 0.14.0 ◆ The language is still pre-1.0 and changes between releases can break builds. Migrating to a newer version means updating the build.zig and adapting to any stdlib changes. The core SIMD and mmap logic is portable, but the build configuration and CLI parsing are tied to the version used here. . The language moves fast and I have not migrated to a newer version yet. The build will break if you try with a different Zig release.
No gzip support. No FASTQ support. No BED. No sub-sequence extraction. The benchmarks show impressive numbers because the tool does one narrow thing and does not worry about the rest. That is honest but it is also limited.
The Rust ecosystem has several FASTA indexing libraries. rust-htslib wraps htslib and provides FASTA indexing through it. needletail is a streaming FASTA/Q parser with speed claims. Both are API libraries, not CLI tools. I chose not to deal with Cargo build complexity and Rust's CLI tooling ecosystem for what I wanted as a learning project. That is a personal constraint, not a technical judgment.
What is next
The current v0.1.0 only indexes. The repository ◆ github.com/eneskemalergin/z-fasta. The README has a more detailed roadmap. already has a roadmap for what comes after:
z-fasta get- O(1) sub-sequence extraction by name or region. The other half ofsamtools faidx.z-fasta bed- Extract sequences for every entry in a BED file in a single pass.z-fasta digest- In-silico trypsin digestion. If the scanner already moves through FASTA at memory bandwidth, computing peptide masses during the scan is a natural extension.- Gzip support - Requires a decompression library. I have not committed to the complexity yet.
These exist as plans, not code. The tool is fast at indexing. It needs to be useful at more than that before it replaces anything in a real pipeline.
Where I think this can go
z-fasta is a small tool that does one thing correctly and fast. It is also the first step in a larger idea: a suite of high-performance bioinformatics utilities in Zig. The opinion pieces I have been writing argue that Zig fits the boring foundation layer: parsers, indexers, validators. z-fasta is the first proof that the performance argument holds.
It is not a replacement for samtools. Not yet. Maybe not ever. It is a demonstration that a small, focused tool in a systems language can beat a mature, general-purpose tool on its own turf. Whether that matters depends on whether the rest of the functionality gets built.
Open source (MIT) at github.com/eneskemalergin/z-fasta.