Research
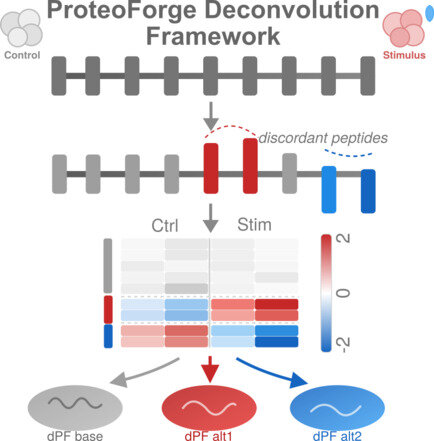
Standard protein-level quantification hides biologically important variation at the proteoform level, and existing deconvolution methods break down when missing values are common. ProteoForge addresses this with an imputation-aware statistical model that identifies peptides behaving discordantly from their parent protein, clusters co-varying peptides, and constructs differential proteoforms (dPFs). The framework consists of four modules: data processing and normalization, discordant peptide identification, quantitative clustering, and dPF construction. Benchmarking on simulated and spike-in datasets showed that ProteoForge maintains high accuracy and stability even under heavy missingness, outperforming existing deconvolution approaches. Applied to a lung cancer hypoxia dataset, ProteoForge uncovered extensive proteoform-level regulation that conventional protein-level analysis missed entirely.
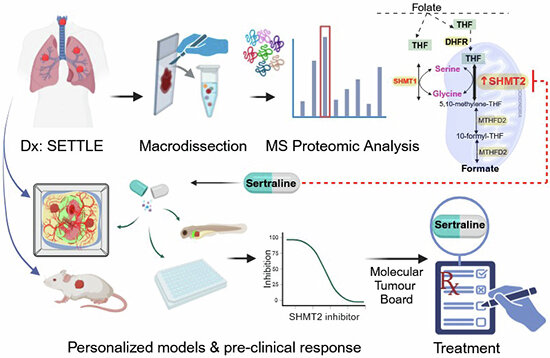
Translating precision oncology into effective therapies for hard-to-cure childhood malignancies remains a major challenge. This study presents a case for combining proteomics with patient-derived xenograft (PDX) models to identify personalized treatment within a clinically actionable window. For an adolescent with a progressive spindle epithelial tumor with thymus-like elements (SETTLE), proteomics identified elevated SHMT2 as a targetable protein within two weeks of biopsy, pointing to the antidepressant sertraline as a candidate therapy. PDX models grown from the patient's tumor confirmed drug sensitivity. The work demonstrates that proteomics can complement genomics in real-time pediatric oncology, delivering actionable molecular targets faster than genome-only approaches.

This thesis develops computational and statistical methods for proteoform-level analysis of bottom-up mass spectrometry data in childhood cancer. Traditional proteomics workflows focus on protein-level readouts, but post-translational modifications and proteoforms carry biologically critical information that protein-level summaries obscure. The work introduces the QuEStVar equivalence testing framework for identifying quantitatively stable and variable proteins across biological systems, and the ProteoForge framework for imputation-aware differential proteoform discovery. Together, these methods shift proteomics analysis from protein-level to peptide-level and fragment-level granularity, enabling detection of regulatory events invisible to conventional approaches. Applications span pediatric leukemia proteomes, cancer cell line panels, and hypoxia-driven proteoform regulation.
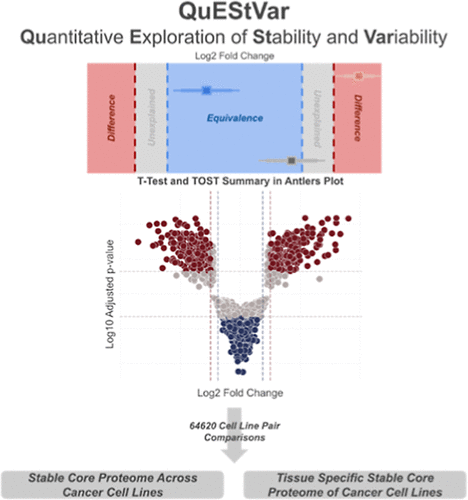
Most proteomics analyses focus on what changes between conditions, but knowing what stays the same is equally important. QuEStVar (Quantitative Exploration of Stability and Variability) combines differential testing with two-one-sided t-tests (TOST) for equivalence, classifying each protein as statistically different, equivalent, or indeterminate. Applied to a panel of 360 cancer cell lines spanning 25 tissue types, QuEStVar identified a conserved core proteome whose stable proteins are enriched in transcription, translation, and nucleocytoplasmic transport. These functional modules define the shared molecular machinery that cancer cells maintain regardless of tissue origin or subtype, offering a new lens for studying biological systems through what is preserved rather than what is disrupted.
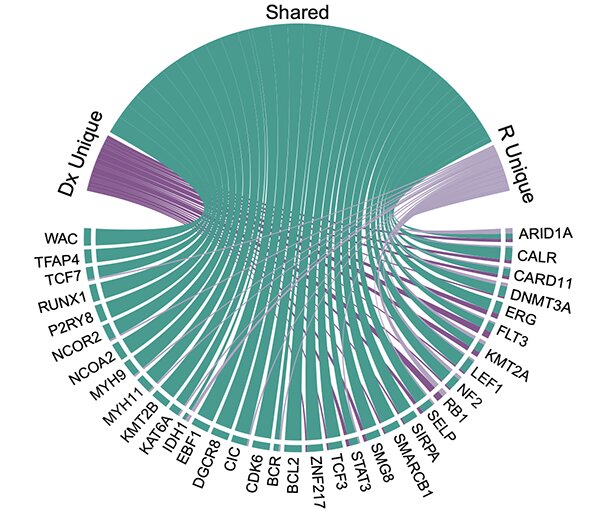
Childhood ALL relapses often arise from subclonal outgrowths, but the impact of clonal evolution on the actionable proteome and drug response is poorly understood. This study presents a comprehensive retrospective analysis of paired diagnosis and relapse ALL specimens using targeted next-generation sequencing and quantitative proteomics. The results show that actionable genome variants and proteomes remain largely stable through disease progression, and paired viably frozen biopsies confirm high correlation of drug response to variant-targeted therapies. Proteome analysis further prioritized PARP1 as a pan-ALL target candidate required for survival following cellular stress, with both diagnostic and relapsed samples showing robust sensitivity to PARP1/2 inhibitors. The findings support initiating precision oncology approaches at diagnosis and incorporating proteome analysis to determine therapy sensitivities likely retained at relapse.

Comprehensive analysis of proteomics data across protein, peptide, and post-translational modification (PTM) levels remains inaccessible to most biologists. SQuAPP is an R/Shiny web application that provides a streamlined workflow for quality control, data preprocessing, statistical analysis, and visualization of quantitative proteomics data. It processes protein, peptide, and PTM datasets in parallel, then integrates them quantitatively. This parallel processing enables rapid identification of protein-level-independent modulation of modifications and intuitive interpretation of dynamic dependencies between different PTMs. SQuAPP is available as an online app, a local R installation, and a Docker container, making it accessible to both expert and novice users without requiring programming.
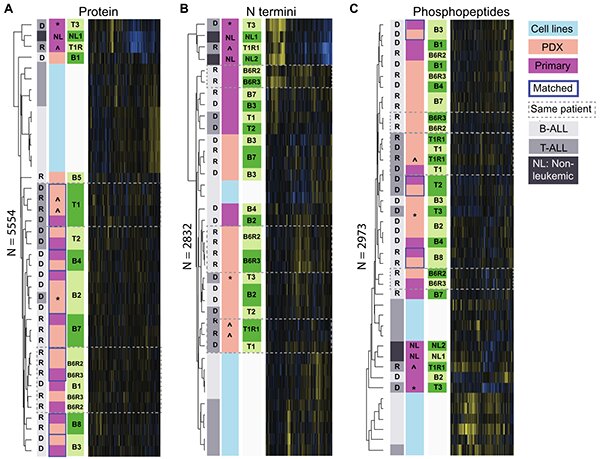
Murine xenografts of pediatric leukemia faithfully recapitulate genomic aberrations, but whether this fidelity extends to the functional proteome was unclear. This study performed a multi-level proteomic comparison of 11 pediatric B- and T-cell ALL patients and 19 corresponding xenograft models, examining global protein abundance, phosphorylation, and proteolytic processing. The results show that PDX models broadly reflect the patient proteome landscape, preserving the majority of disease-relevant protein expression patterns. However, select pathways diverge between patient and xenograft, highlighting specific biological contexts where preclinical xenograft data should be interpreted with caution. Targeted next-generation sequencing confirmed retention of key genetic abnormalities across the matched pairs.
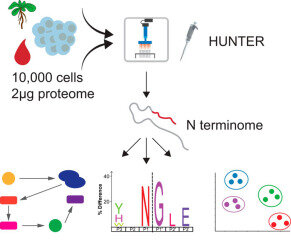
Protein N termini are definitive markers of truncated, alternatively translated, or modified proteoforms, but existing enrichment methods require large sample amounts that exclude many biologically relevant contexts. This paper introduces HUNTER (High-efficiency Undecanal-based N Termini EnRichment), a scalable and automatable method for N-terminal peptide enrichment from microscale samples. HUNTER identified over 1,000 N termini from as little as 2 micrograms of HeLa cell lysate, opening proteoform-level analysis to previously inaccessible sample types. The method enables proteome-wide, unbiased identification of site-specific regulatory proteolytic processing and protease substrates in samples where time- and space-confined proteolytic events would otherwise go undetected.
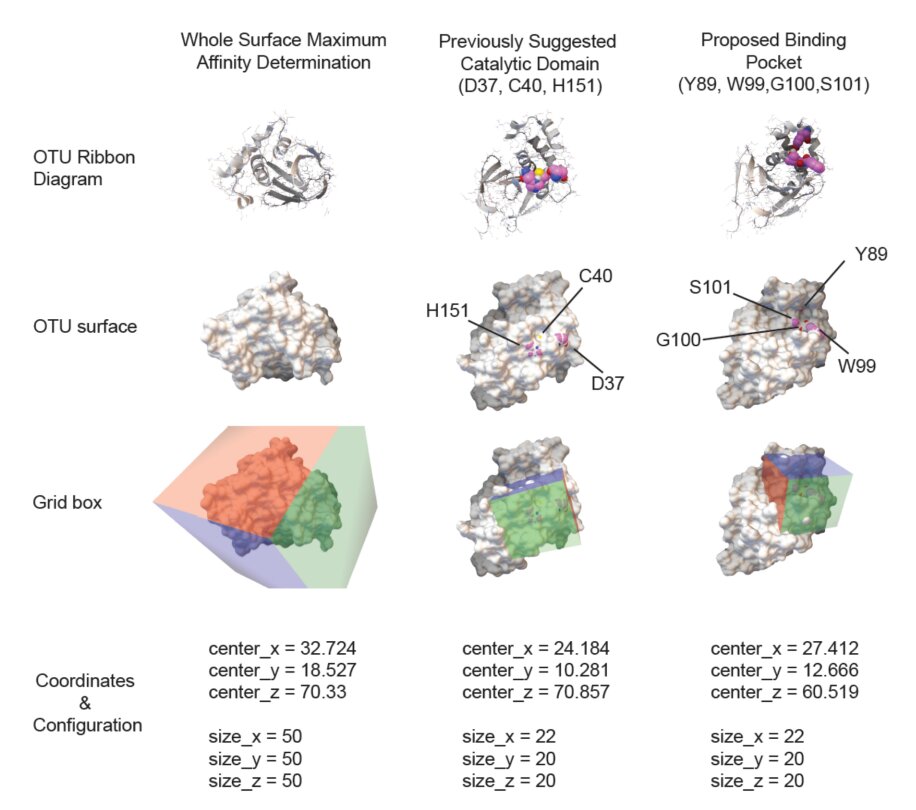
Crimean-Congo hemorrhagic fever virus (CCHFV) is a tick-borne pathogen with high fatality rates and no approved treatments. The viral OTU protease plays a key role in immune evasion by stripping ubiquitin and ISG15 from host signaling proteins, making it an attractive drug target. This study used molecular docking and virtual screening to map the OTU protease structure and identify small molecule binding pockets capable of inhibiting its activity. Compound libraries were screened against the protease to rank candidate inhibitors by predicted binding affinity and pocket geometry. The work provided early structural insight into druggable sites on the CCHFV OTU domain, laying groundwork for future antiviral development against this neglected pathogen.
Characterization of a New Type of Neoantigen in T-Cell Acute Lymphoblastic Leukemia (T-ALL) by Cell Surface Terminomics
Proteomics Workshop: Common Downstream Analysis Pipeline
Exploring Stable Proteome in Cancer Cell Line Atlas with QuEStVar
InPACCT: Integrated Proteomics Analysis of Curated Childhood Tumours
InPACCT: Integrated proteomics analyses of curated childhood tumours
Stable Proteome in Cancer Cell Lines with Combined Testing
Insightful, Integrated Analyses of Public Pediatric Cancer Proteomics with InPACCT
Normalization in Proteomics: Past, Present, and Future
Sensitive determination of proteolytic proteoforms in limited samples
Statistical methods for proteomics data
Mathematics and Computer Science
COMP 3317: Algorithms
COMP 3320: Programming Languages
COMP 3322: Software Engineering
COMP 2415: Systems Programming
Introductory Python Programming