mzarc v0.0.1: Can Domain-Specific Compression Beat General-Purpose Codecs on Mass Spectra?
TLDR: mzarc is an early prototype of a domain-specific compression codec for mass spectrometry data in Zig. On one DDA dataset, lossless .mzv1 compresses to 27.89 MiB from 75.55 MiB mzML, beating gzip and trailing mzMLb by 11.64 MiB. Lossy at q=4096 hits 13.17 MiB with 0.218% p95 relative intensity error. Decode throughput is 167 MiB/s, roughly 27x faster than mzMLb. This is v0.0.1. One dataset. Scalar codec only. The remaining lossless size is almost entirely the exact m/z stream.
Why this exists
I have been thinking about compression for mass spectrometry data since before I started learning Zig. The opinion pieces I wrote over the past few months keep circling the same idea: storage is an unaccounted cost, open formats inflate file sizes, and the tools to fix it exist but nobody adopts them.
At some point I decided to stop writing about it and start building.
mzarc is a question expressed as a codebase: can a codec that knows about mass spectra beat general-purpose compressors, and can it do it with decode speed fast enough to feed a search engine directly?
This is v0.0.1. The answer so far is partial. I am sharing it because the partial answer is interesting, and because the honest version of an early experiment is more useful than a polished launch.
What this is
mzarc is a domain-specific, asymmetric compression codec for mzML-derived mass spectrometry spectra. Asymmetric means encode can be slow. Decode must be fast.
The pipeline right now is deliberately narrow:
mzML -> Python dump tool -> flat binary dump -> Zig codec -> .mzv1 filePython handles mzML ingestion once. Zig handles the transform and decode repeatedly. This keeps XML parsing out of the codec work. The binary dump is an internal handoff format, not a format anyone should use directly.
The codec stack itself is four scalar transforms composed in sequence:
- Quantize: m/z to fixed-point; intensity to log-scale at configurable q.
- Delta: intra-spectrum delta coding on sorted m/z arrays.
- FOR bitpack: frame-of-reference packing with per-spectrum bit widths.
- Block: 128 spectra per block, CRC32 validated, MS1 and MS2 in separate block streams.
That is the whole thing. No entropy coding yet. No SIMD. No cross-spectrum delta. Those come later, if the scalar baseline justifies them.
What I measured
One dataset: 15HCD_1 ◆ From PXD075509. 9001 spectra, 2,668,458 total peaks, 917 MS1, 8084 MS2. DDA acquisition on a Thermo instrument. . One machine. Ten repeat runs per operation. Benchmarked against mzMLb, MScompress, gzip, and zstd. All tools got the same dump as input so the comparison isolates the codec, not the XML parsing.
Size
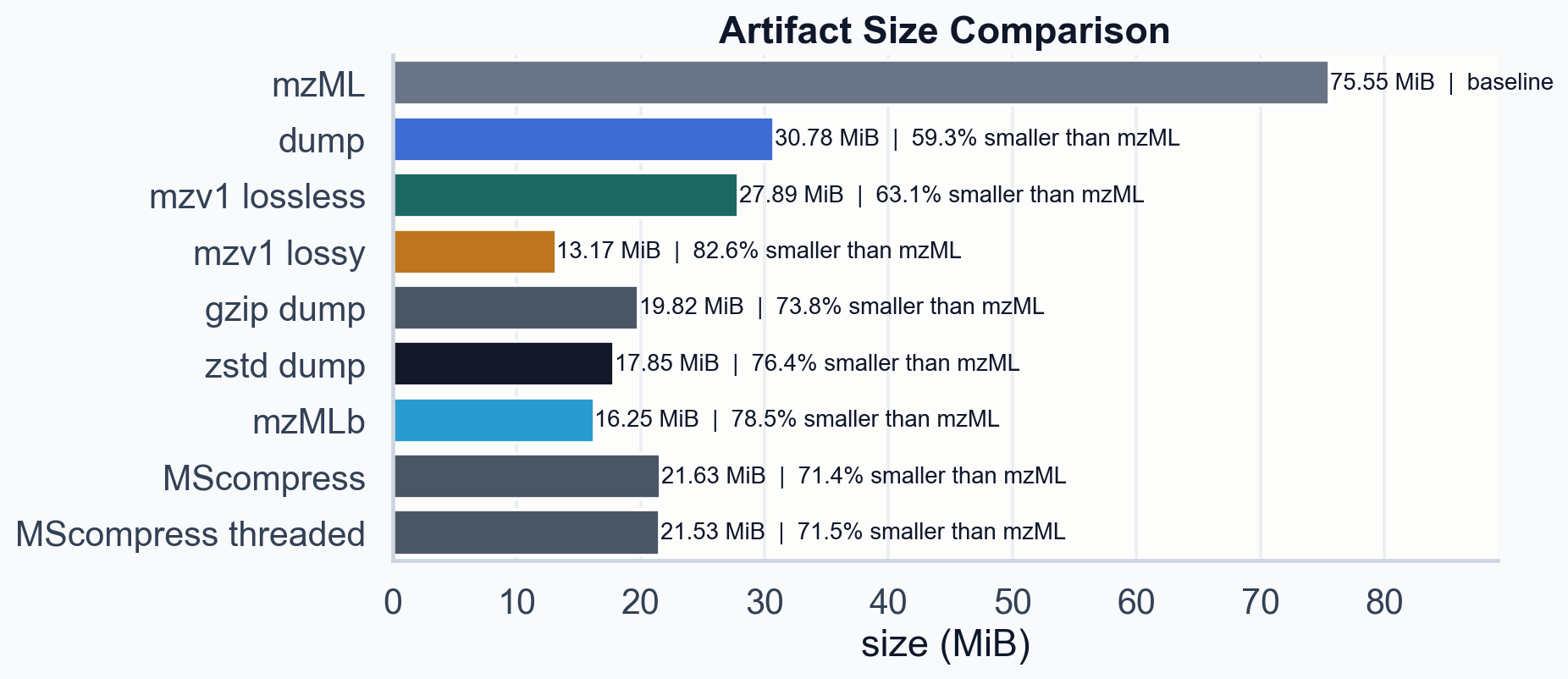
File size comparison on 15HCD_1. Lossless mzv1 (27.89 MiB) beats gzip (19.82 MiB) and the dump itself (30.78 MiB). Lossy at q=4096 (13.17 MiB) is smaller than mzMLb (16.25 MiB).
| Artifact | Size | vs mzML | vs dump |
|---|---|---|---|
| :--- | ---: | ---: | ---: |
| mzML | 75.55 MiB | 100% | 245% |
| dump (binary flat) | 30.78 MiB | 41% | 100% |
| mzv1 lossless | 27.89 MiB | 37% | 91% |
| mzv1 lossy q=4096 | 13.17 MiB | 17% | 43% |
| gzip dump | 19.82 MiB | 26% | 64% |
| zstd dump | 17.85 MiB | 24% | 58% |
| mzMLb | 16.25 MiB | 22% | 53% |
| MScompress | 21.63 MiB | 29% | 70% |
The lossless path clears a low bar: it beats gzip and is smaller than the internal dump itself. That means the transforms are doing real work, not just shuffling bytes. It trails zstd on the dump and mzMLb, which is expected. mzMLb uses HDF5 with blosc:zstd compression. mzarc v0.0.1 uses scalar FOR bitpacking with no entropy coding. The gap is roughly 11.64 MiB, and most of it is in one place ◆ The m/z stream. 17.50 MiB of the 27.89 MiB lossless file. That single stream is larger than the entire mzMLb file (16.25 MiB). If entropy coding can cut it in half, the lossless path wins. .
Where the bytes go
| Stream | Lossless | Lossy q=4096 |
|---|---|---|
| :--- | ---: | ---: |
| Structural | 0.04 MiB (0.1%) | 0.04 MiB (0.3%) |
| Spectrum metadata | 0.17 MiB (0.6%) | 0.17 MiB (1.3%) |
| m/z stream | 17.50 MiB (62.8%) | 9.17 MiB (69.6%) |
| Intensity stream | 10.18 MiB (36.5%) | 3.80 MiB (28.8%) |
The m/z stream is 17.50 MiB in lossless mode. That is 62.8% of the total file. The intensity stream shrinks from 10.18 MiB to 3.80 MiB under lossy quantization, exactly as designed. The m/z stream barely moves between lossless and lossy because the current quantizer preserves m/z exactly in both paths. Fixing this requires either lossy m/z quantization with controlled bounds, or an entropy coding layer that compresses the delta-encoded m/z residuals. Both are on the list. Neither is in v0.0.1.
Throughput
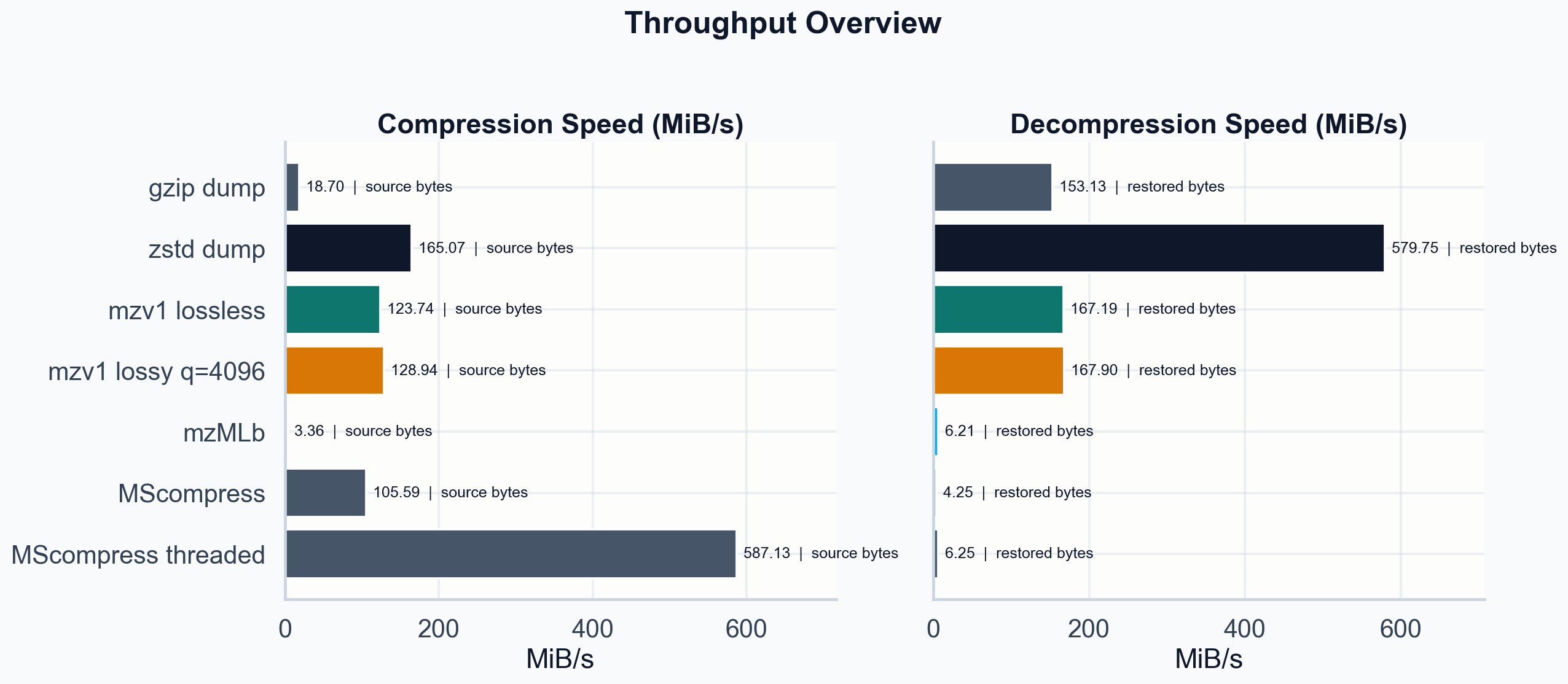
Throughput in MiB/s. mzv1 encode and decode both exceed 120 MiB/s. mzMLb decode (6.2 MiB/s) is 27x slower.
| Operation | Throughput | Time |
|---|---|---|
| :--- | ---: | ---: |
| mzv1 lossless encode | 123.7 MiB/s | 0.25s |
| mzv1 lossless decode | 167.2 MiB/s | 0.18s |
| mzv1 lossy encode | 128.9 MiB/s | 0.24s |
| mzv1 lossy decode | 167.9 MiB/s | 0.18s |
| mzMLb encode | 3.4 MiB/s | 22.5s |
| mzMLb decode | 6.2 MiB/s | 5.0s |
| MScompress encode | 105.6 MiB/s | 0.72s |
| MScompress decode | 4.3 MiB/s | 7.2s |
| zstd dump decode | 579.7 MiB/s | 0.05s |
mzv1 decode at 167 MiB/s is 27x faster than mzMLb decode. It is slower than zstd on the dump, which is expected: zstd has years of SIMD-optimized C. The scalar Zig codec has none. The question is whether the gap closes with entropy coding and SIMD, or whether general-purpose compressors will always be faster on decode. I do not know yet.
MScompress decode at 4.3 MiB/s is the slowest path in the benchmark. Its threaded encode hits 587 MiB/s, which is impressive, but the asymmetry is in the wrong direction for a format designed to be decoded many times.
How to reproduce these benchmarks
Run from the repository root:
uv run python tools/benchmark_v1.py \
--repeats 10 \
--external-baselines mzmlb,mscompress \
--mscompress-benchmark-threaded \
data/PXD075509/15HCD_1.mzML
Output goes to benchmark/report.json and benchmark/report.md. Plots land in benchmark/plots/. The data shown here is from commit 93459bd5.
Fidelity
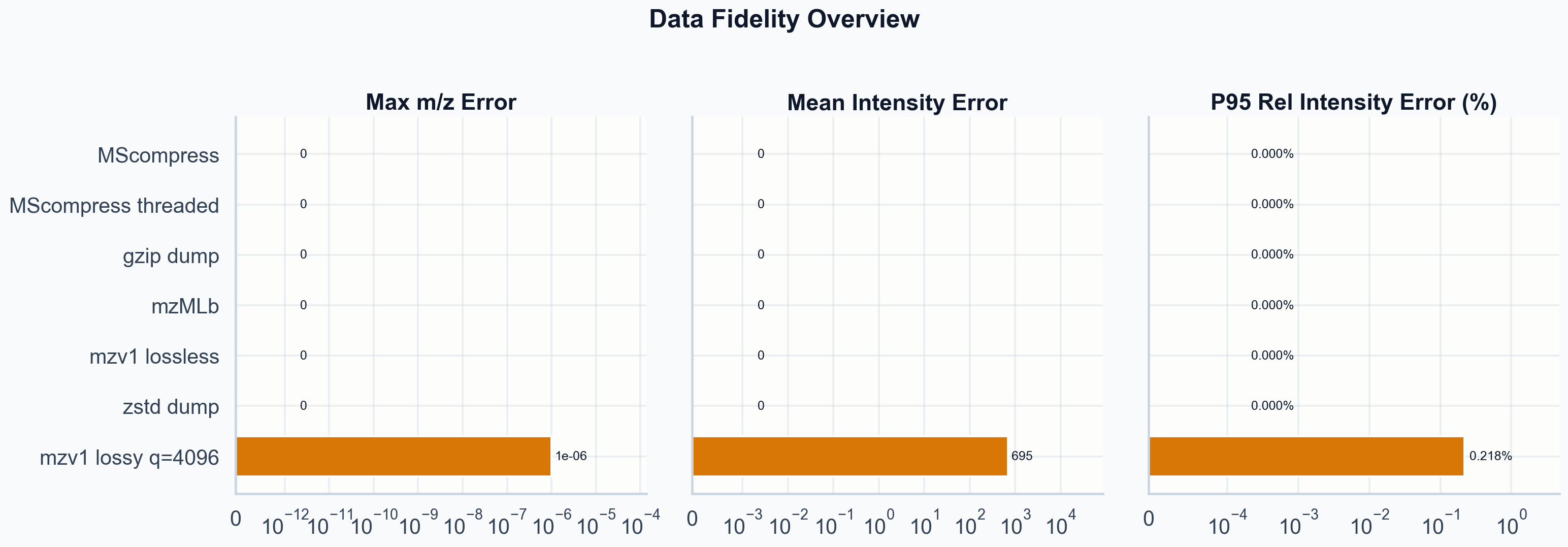
Fidelity overview. Lossless paths are exact. Lossy q=4096 shows controlled intensity error with m/z error at the ppm level.
Lossless mzv1 round-trips exactly. Every m/z value and every intensity survives encode-decode unchanged ◆ This is tautological by design. A lossless codec that changed data would be a bug. The claim matters only because it separates the codec correctness from the quantization question, which is where the interesting tradeoffs live. . So does the original scan order.
Lossy at q=4096: max absolute m/z error is 1.0e-06 Da. Mean absolute intensity error is 695 (raw counts). P95 relative intensity error is 0.218%. P99 is 0.238%. These are controlled errors within the quantization bounds. The lossy tradeoff sweep makes this explicit:
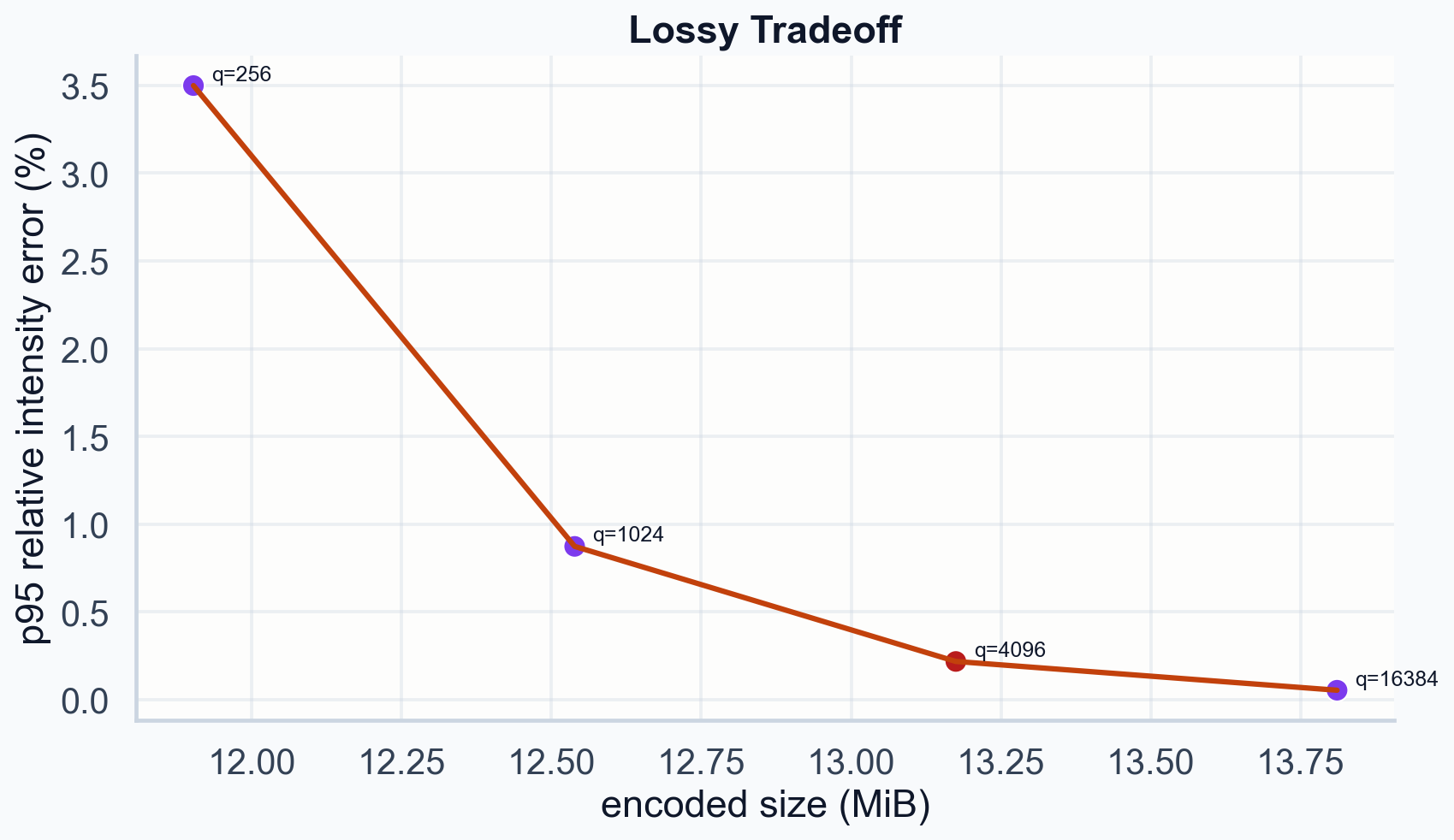
Lossy tradeoff. Higher q preserves more precision at modest size cost. q=16384 gives p95 error of 0.055% at only 0.64 MiB more than q=4096.
| q | Size | P95 rel intensity error | P99 rel intensity error |
|---|---|---|---|
| ---: | ---: | ---: | ---: |
| 256 | 11.90 MiB | 3.499% | 3.813% |
| 1024 | 12.54 MiB | 0.874% | 0.950% |
| 4096 | 13.17 MiB | 0.218% | 0.238% |
| 16384 | 13.81 MiB | 0.055% | 0.059% |
At q=16384 the p95 error is 0.055%. The file is 0.64 MiB larger than q=4096. For archival storage, that cost is near zero.
What I did not measure
Search impact. This is the measurement that matters most. Do peptide identifications and FDR estimates change after a round-trip through lossy mzv1? The benchmark tracks numeric fidelity. It does not track downstream biological conclusions. That requires running a search engine on the original and round-tripped spectra and comparing the results. I have not done that yet.
Assumptions
Several assumptions are baked into v0.0.1 that may turn out to be wrong:
- One dataset is representative. 15HCD_1 is DDA on a Thermo instrument. DIA data looks different. timsTOF data looks different. Profile-mode data is an order of magnitude larger. Every conclusion in this post is conditional on one file.
- The dump is a fair baseline. Stripping XML overhead is an obvious first step. It is not a format. The dump baseline shows how much of the size reduction is just removing interchange overhead vs actual compression. The gap between mzML (75.55 MiB) and the dump (30.78 MiB) is the XML tax. The gap between the dump and mzv1 lossless (30.78 to 27.89 MiB) is the codec doing real work. That gap is 2.89 MiB. It is real. It is small.
- Scalar FOR is enough. The current size is dominated by the exact m/z stream. Entropy coding (rANS, tANS) should shrink that stream significantly. Cross-spectrum delta should help on DIA where consecutive spectra share precursors. Both are unimplemented. If they do not close the gap to mzMLb, the thesis is in trouble.
- Python is an acceptable dependency. The prototype ingests mzML through pyteomics. This is fine for benchmarking. It is not fine for production. A native Zig mzML reader is on the roadmap. It is not in v0.0.1.
- Decode speed matters more than encode. This is the asymmetric design bet. Encode happens once per file. Decode happens every time a search engine reads the data. If decode is not fast enough to feed a search engine without becoming the bottleneck, the format is not useful.
Limitations
This is v0.0.1. The list of things not yet done is longer than the list of things done:
- One dataset. No DIA. No timsTOF. No multi-instrument validation.
- No entropy coding. The m/z stream is delta-encoded and FOR-packed but not entropy-coded. That is the largest remaining compression opportunity.
- No SIMD. The decode path is scalar. SIMD FOR unpack should roughly double decode throughput.
- No cross-spectrum delta. Consecutive DIA spectra share precursors. Encoding differences between spectra rather than absolute values should reduce the m/z stream significantly.
- No search impact measurement. Numeric fidelity is not the same as biological fidelity.
- No native mzML reader. Python dependency for ingestion.
- No comparison against MS-Numpress. It is the most natural baseline for array-level compression inside mzML and should be added to the benchmark.
What comes next
The immediate next steps are narrow:
- Fix the m/z stream. Entropy coding first. RANS or tANS. If the m/z stream shrinks from 17.50 MiB to something closer to 5-7 MiB, the lossless path beats mzMLb. That is the threshold that decides whether to keep going.
- Add a second dataset. DIA on a different instrument. If the codec assumptions break on DIA data, that is better learned now than after months of optimization.
- Measure search impact. Run MSFragger or DIA-NN on original and round-tripped spectra. If peptide IDs and FDR are unchanged at q=16384, the lossy path is viable. If they drift at any q, the quantization scheme needs revision.
After that, the project either has evidence to continue or evidence to stop.
Where this fits
mzarc is the second Zig tool I have shipped ◆ The first was z-fasta, a FASTA indexer that runs 9-17x faster than samtools. I wrote about it here. . z-fasta proved that a single static binary could beat established tools on a narrow, well-defined problem. mzarc is trying to prove something harder: that domain-specific encoding can beat general-purpose compression on a format that matters to my field.
The honest assessment after v0.0.1 is that the thesis is not yet proven. The lossless path trails mzMLb. The decode path is fast but scalar-only. The dataset coverage is one file. The search impact is unmeasured.
But the architecture is sound. The codec composes cleanly. The transform chain round-trips exactly. The benchmark pipeline is reproducible. The byte accounting points directly at the remaining gap. These are good foundations.
The next version will either close the gap to mzMLb or explain why it cannot. Either outcome is useful.
Open source (MIT) at github.com/eneskemalergin/mzarc.